RFE on Breast Cancer Dataset
Datasets
- TCGA-BRCA 美國癌症基因體圖譜計畫
- GSE66313 高通量基因表達數據庫
Description
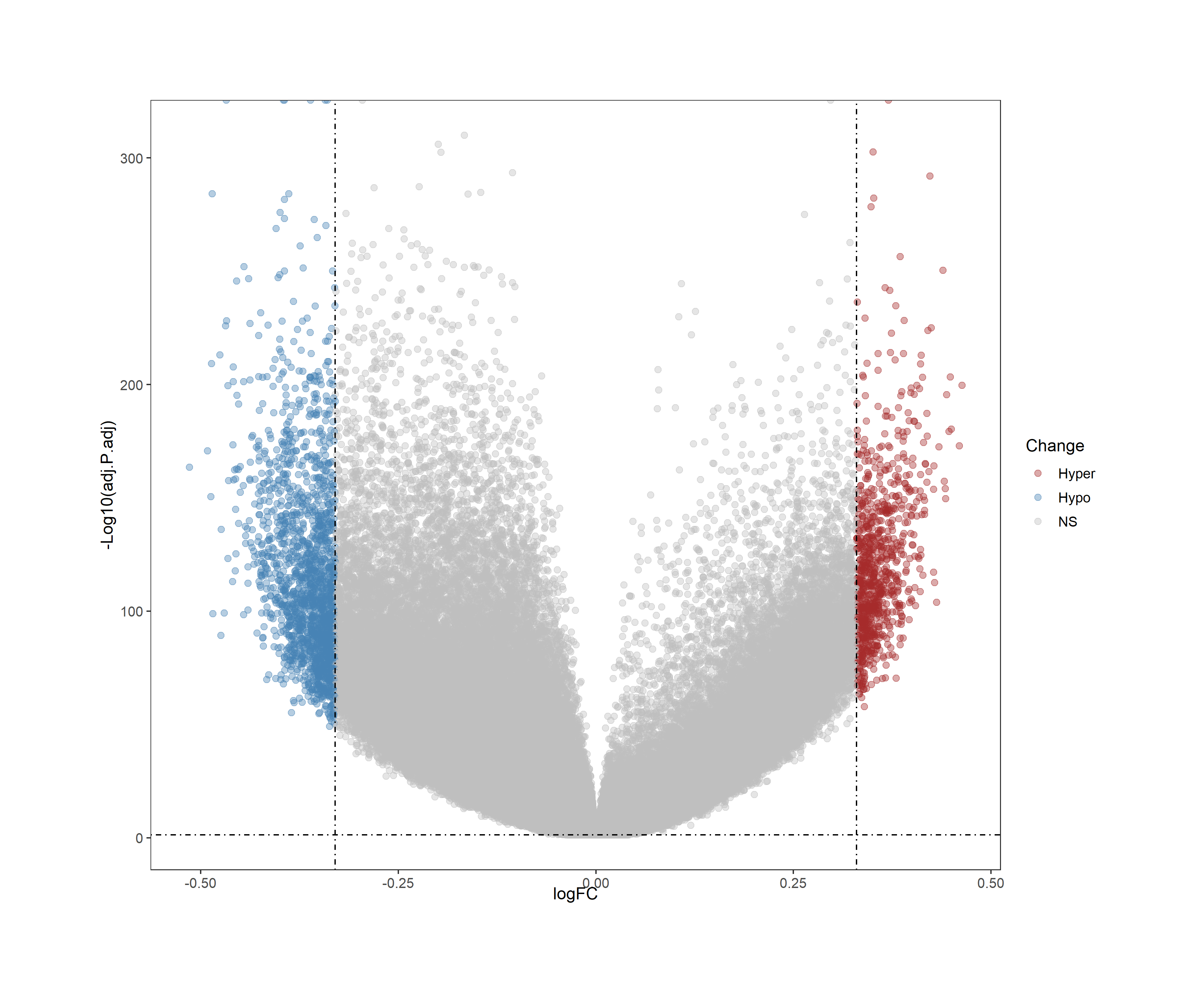
By using ChAMP package, a volcano plot can be generated to visualize the differentially methylated regions (DMRs) in the TCGA-BRCA dataset. The volcano plot displays the relationship between the significance (p-value) and the magnitude of change (fold change) of the DMRs.
Dbeta Calculation and DMR Filtering
Extensive preprocessing of GSE66313 and TCGA-BRCA methylation data was performed using the ChAMP package in R. This involved several critical steps:
- Quality Control: Removal of low-quality probes, SNP-related probes, and cross-reactive probes
- Normalization: BMIQ normalization to adjust for probe type bias (Infinium I vs II)
- Batch Effect Correction: ComBat algorithm was applied to remove potential batch effects
- Differential Methylation Analysis: Calculated differentially methylated beta values (Dbeta) between tumor and normal tissues
- Statistical Filtering: Applied significance threshold of p < 0.05
- Annotation: DMRs were mapped to genes based on their genomic locations and proximity to TSS (Transcription Start Sites)
The resulting differentially methylated regions (DMRs) represent critical epigenetic alterations that may drive breast cancer development and progression. Positive Dbeta values indicate hypermethylation in cancer tissues, while negative values represent hypomethylation.
Hyper/Hypo Methylation Distribution - TCGA-BRCA
Feature Distribution - TCGA-BRCA
Hyper/Hypo Methylation Distribution - GSE66313
Feature Distribution - GSE66313
Principal Component Analysis (PCA)
PCA of TCGA-BRCA
PCA of GSE66313
Joined Data Analysis
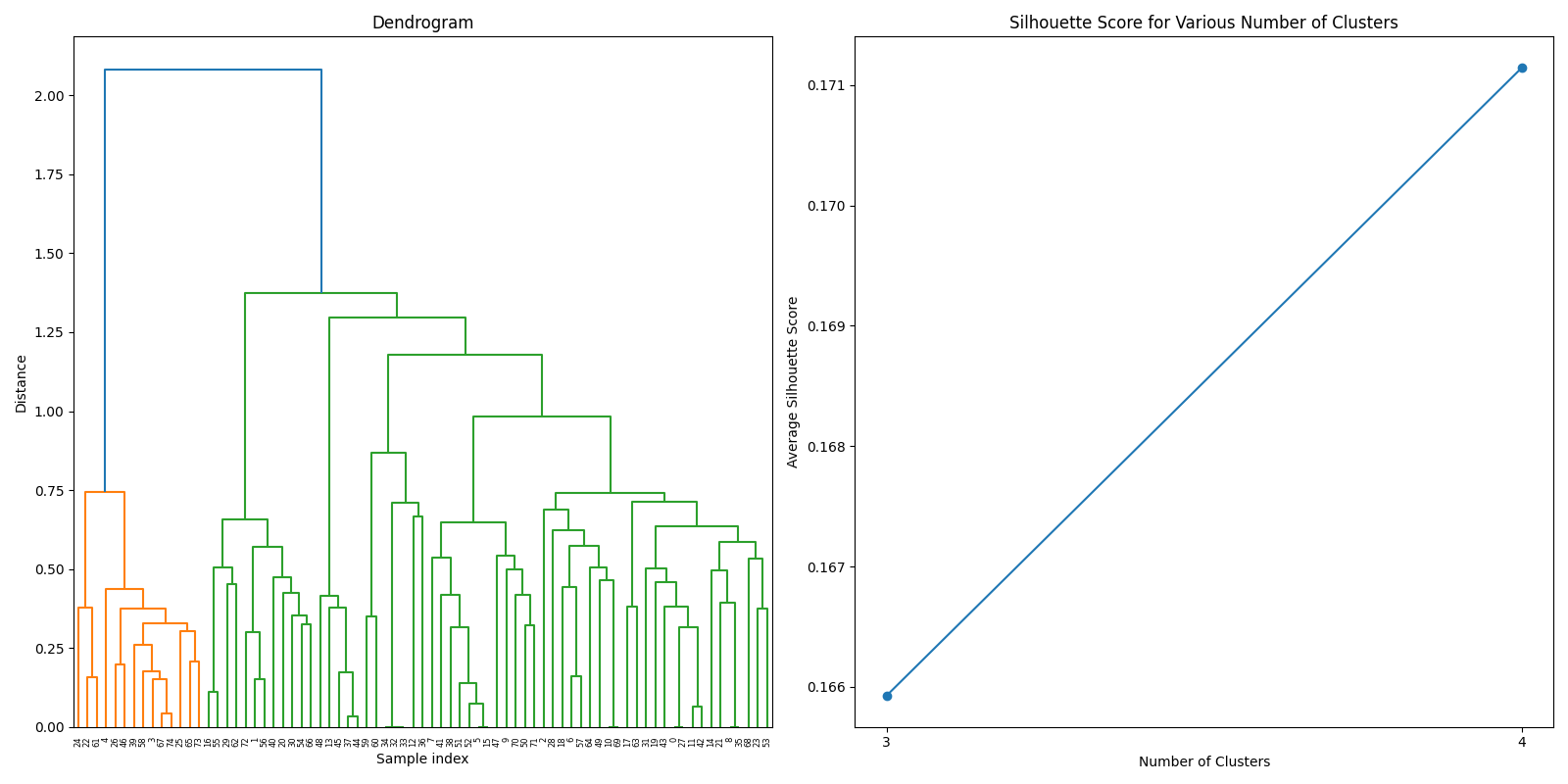
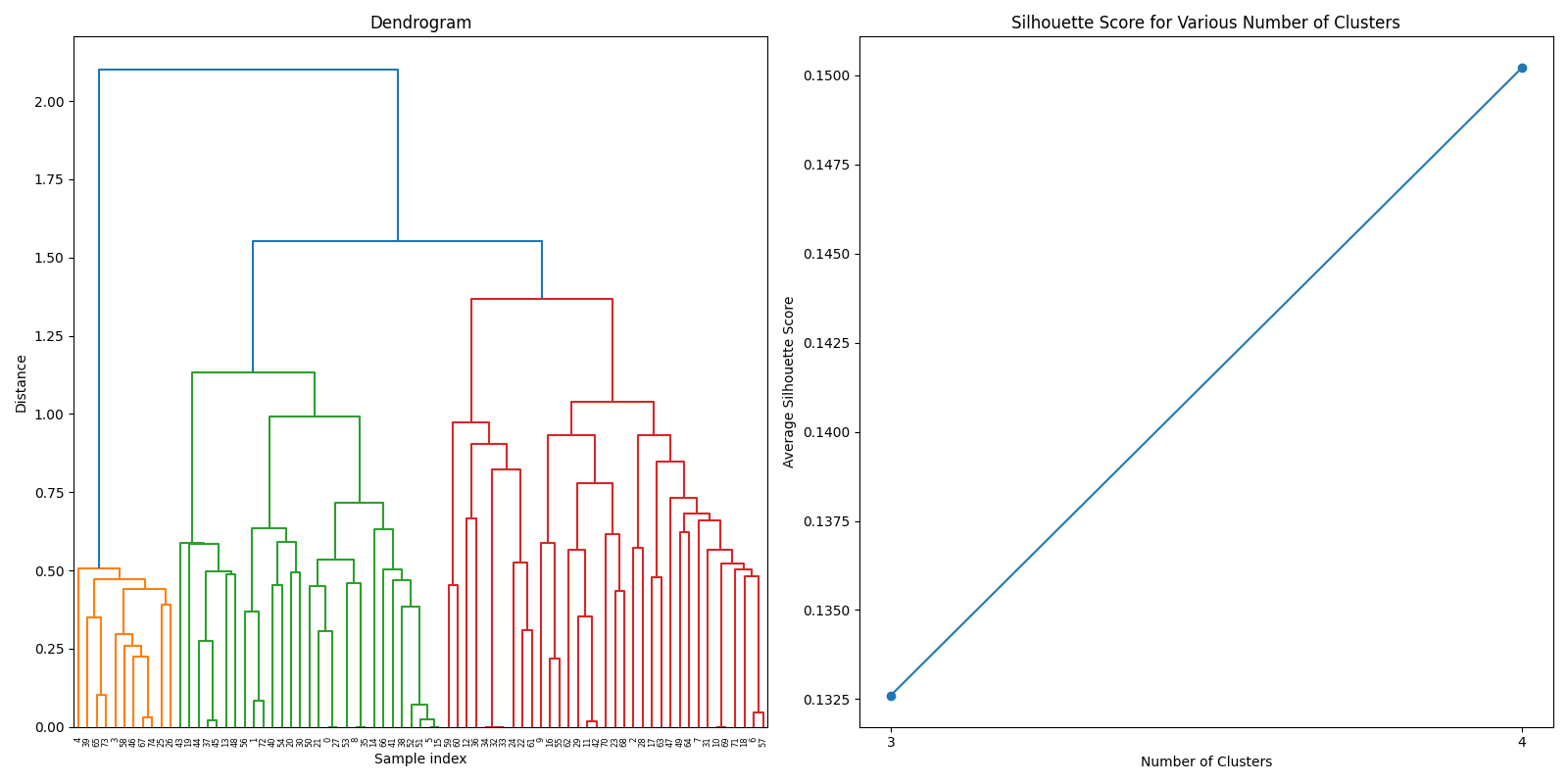
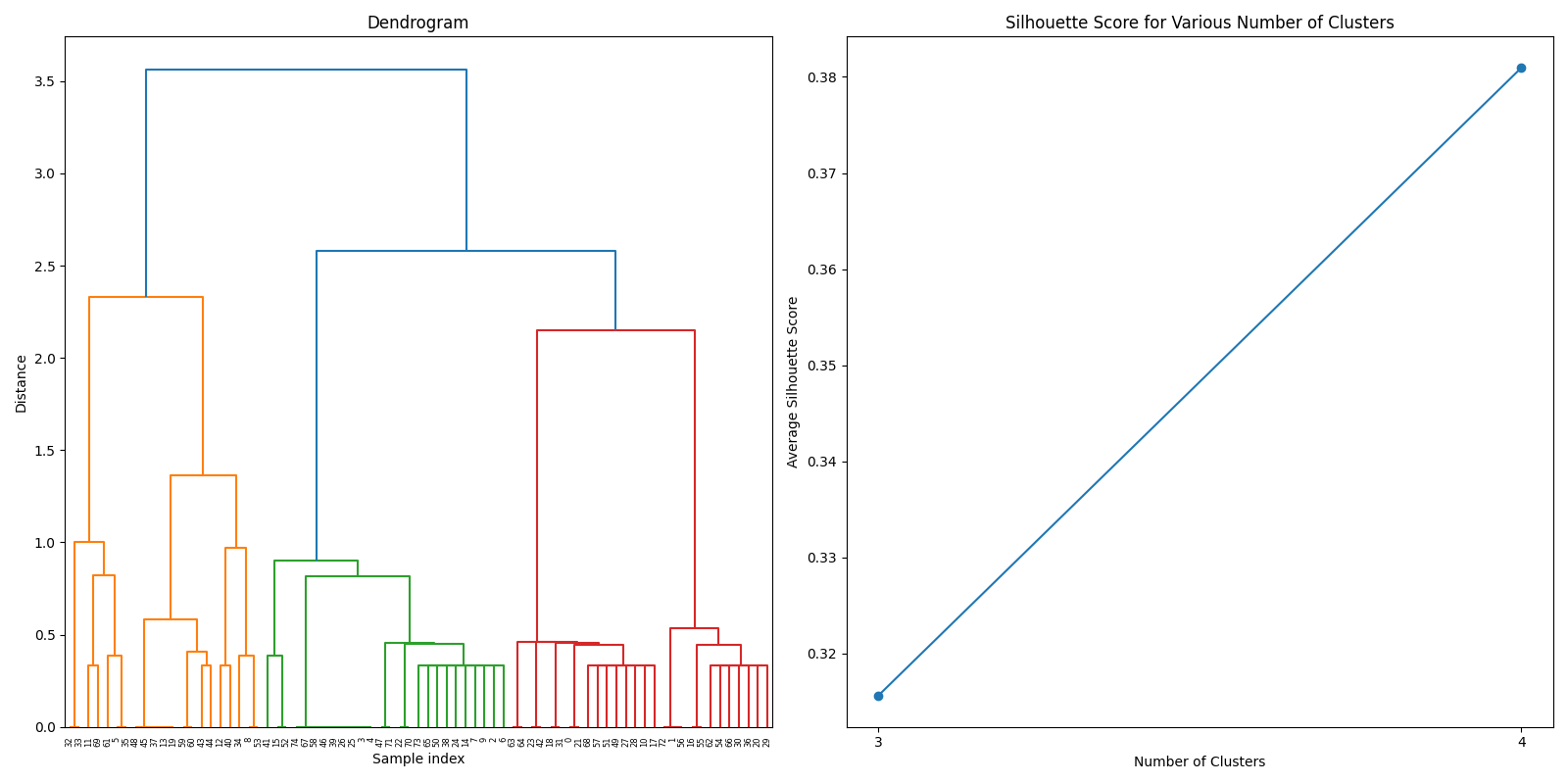
After filtering the DMRs from both datasets, we joined the two datasets to create a comprehensive dataset for further analysis. The joined dataset contains a total of 88 gene, whose similarity was calculated using GO (Gene Ontology) terms. The GO terms were obtained from the Ensembl database, and the similarity was calculated using the GOSemSim package in R.
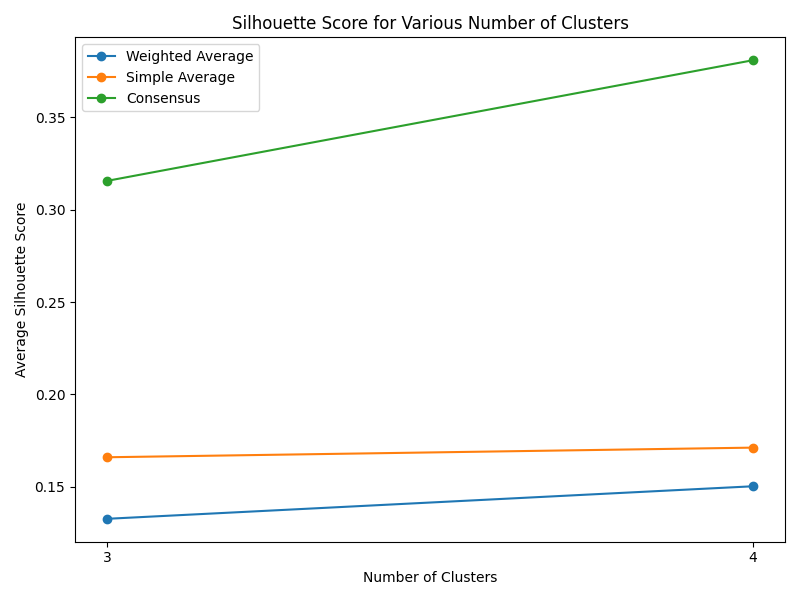
Machine Learning
After thorough evaluation of clustering methodologies, consensus clustering emerged as the superior approach for analyzing our joined dataset due to its robustness in handling gene expression patterns. For biomarker identification, we implemented Recursive Feature Elimination (RFE) with cross-validation to systematically select the most predictive gene signatures while minimizing redundancy. This process ranked genes based on their discriminative power between cancer and normal tissue samples.
The candidate biomarkers underwent rigorous validation through an ensemble-based voting classifier architecture, which integrated predictions from multiple base learners (Random Forest, SVM, and Gradient Boosting) to improve classification stability. To enhance generalization capabilities and address potential overfitting concerns, we employed bootstrap aggregating (bagging) techniques with out-of-bag error estimation. This comprehensive machine learning pipeline delivered robust biomarker combinations with exceptional predictive performance across multiple validation datasets.
Top Gene Combinations with Highest Performance
The table below shows the gene combinations that achieved the highest classification performance:
| Gene Set | Gene 1 | Gene 2 | Gene 3 | Gene 4 | Accuracy | Recall | Specificity | Precision | F1 Score | AUC |
|---|---|---|---|---|---|---|---|---|---|---|
| Set 1 | CFTR | CNTNAP5 | PFN3 | SLC2A10 | 0.932 | 1.000 | 0.864 | 0.880 | 0.936 | 0.997 |
| Set 2 | CNTNAP5 | GNG4 | RGS22 | SLC2A2 | 0.932 | 1.000 | 0.864 | 0.880 | 0.936 | 0.996 |